Graphical representations of rank-2 tensors
[This page uses MathJax. If the Math below doesn’t display properly, please load
this (older, no longer maintained) version instead.]
Let’s first answer the question ‘What is a tensor’, from the perspective of a
scientist.
The laws of physics are formulated in terms of scalars (rank-0 tensors), vectors
(rank-1 tensors), and so forth. A scalar has a magnitude, and it may have a sign. A
scalar does not depend on the coordinate system used to express physical laws.
Examples are energy or temperature.
A vector, such as velocity ,
has a magnitude and a direction, which tell you how fast a particle moves in the
coordinate system along one of the axis directions (positive or negative). In a two- or
three-dimensional Cartesian coordinate system, the vector must then be written with
two or three components. The vector components depend on the coordinate system
you choose. Furthermore, the vector components must change in a specific way upon
transforming between coordinate systems, such that for a particle with mass
the kinetic
energy ,
for instance, is invariant under coordinate transformations.
(‘’ stands for
the energy of translational motion.) In the previous equation, we have the scalar product
of vector
with itself.
Rank-2, -3, and so forth, tensors are generalizations of this concept. As for vectors,
the components of a tensor with rank 2 or higher must transform in a specific way
upon transforming the underlying coordinate system, such that physical laws
expressed in terms of these tensors do not change.
A rank-2 tensor, say ,
will often show up in physical laws in equations such as
, where
and
are vectors,
and
provides the mathematical recipe for changing vector
into
vector .
Below, we use this idea to express the local magnetic field in NMR spectroscopy (a
vector) in terms of the external field (also a vector). Another example would be the
dipole moment (a vector) induced in a medium or in a molecule by an external
electric field (also a vector). The rank-2 tensor involved in the induced dipole
moment-electric field relationship is called polarizability. The energy (a scalar)
associated with the polarization would be given by an expression such as
, where
is a scalar,
and
and
are rank-2 and rank-1 tensors, respectively.
Before we discuss the details of tensor visualization, let’s look at an example along
with a brief description. The language used in the description is explained in detail
further below. The figure was created with this Mathematica notebook (60 kBytes).
For further details see my Downloads page.
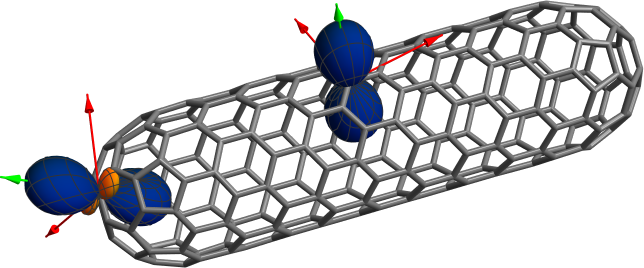
Graphical representations of selected NMR carbon shielding
tensors of a finite (9,0) single-walled carbon nanotube. The blue
and
orange
surfaces indicate the signs and relative magnitudes of the principal components of
the NMR shielding tensors as well as the orientations of the principal axis systems
(PASs) relative to the molecular coordinate frame. The arrows additionally show the
PASs and are color coded (green for positive principal components and red for
negative ones). A sphere would indicate an isotropic tensor. The nanotube shielding
tensors are strongly anisotropic. Data to prepare the figure were taken from
publication [31].
Many optical and spectroscopic properties of molecules that we study in our
research, on a day-to-day basis or occasionally, are rank-2 tensors. Examples are
NMR shielding, NMR spin-spin coupling, electric field gradients at nuclei (giving rise
to the quadrupolar interaction in NMR and other types of spectroscopy),
polarizability, optical rotation, or the magnetic susceptibility. If you know what a
rank-2 tensor is please just scroll down to the detailed description of the
functions that are plotted in the figure shown above. Otherwise, here is a short
explanation:
What is a tensor, and what is a rank-2 tensor? To reiterate, a rank-0 tensor is a
scalar (a single number). A rank-1 tensor is a vector. For a physical quantity defined
in a 3D Cartesian coordinate system such a vector has three components:
A rank-2 tensor can be written as a matrix. For example, for a quantity defined in
a 3D Cartesian coordinate system a rank-2 tensor can be written as (we use
boldface notation for vectors and boldface with a bar for rank-2 tensors here):
|
|
In a nutshell, a tensor of rank greater than zero is a physical quantity that is
represented by more than a single number, the tensor components. These
components transform among each other under a transformation of the coordinate
system such that physical laws written in terms of the tensors remain the same in
different coordinate systems.
For example, the interaction energy (a scalar = rank-0 tensor) between a magnetic
field of
magnitude
(i.e. is the length
of the vector ) and a
magnetic dipole moment
with magnitude
is given by the dot-product of the two vectors:
|
|
The equations assume a Cartesian coordinate system with unit vectors
of length
1. The point is that if we rotate the coordinate system used to describe the vectors
and
(passive
rotation), or if the rotate the two vectors simultaneously in the exact same way (active rotation),
the energy
must remain the same as we have neither changed the angle
between the two vectors nor their lengths. This puts certain constraints on how the
components of the vectors must transform under rotations of the coordinate system
or when they are rotated. Likewise, the components of a rank-2 or higher tensor have
certain transformation rules upon rotations.
The previous equation for
is a good starting point to introduce a rank-2 tensor. If the magnetic
dipole moment is that of an atomic nucleus’ spin, the energy
is quantized
and we can observe transitions between ‘parallel’ and ‘anti-parallel’ relative orientations
of and
by spectroscopy.
The magnetic field
is then the field created by the powerful magnet of an NMR spectrometer. But there
is something else going on: there are electrons around a magnetic nucleus in a
molecule, and there are other atoms around, and there are other molecules around
those. Therefore the magnetic field experienced by the nuclear spin magnetic
moment is not the field generated by the spectrometer magnet but a local field. The
local field may differ in magnitude and orientation from the external spectrometer
field, and it may depend on the relative orientations of the external field and the
molecule. Usually, the magnitude of the local field is reduced relative to the external
field, and the effect is therefore referred to as NMR shielding. But how to describe it
theoretically?
Let’s denote the external spectrometer field by
and the local
field by .
Considering that the local field will usually be smaller in magnitude (hence, one
needs a subtraction of some sort), we can write
|
|
Here, is the nuclear
magnetic shielding tensor. If
were a scalar, then would
always be parallel to ,
and therefore would also
always be parallel to .
This is not general enough to describe experimental observations. Instead, NMR
shielding is a rank-2 tensor:
|
|
The interaction energy between the nuclear magnetic moment and the magnetic
field is
|
|
Since
is a matrix-vector product, the result is a vector and one can take the dot-product
with the magnetic moment vector afterwards to obtain the shielding contribution to
. As in
the simpler case discussed above, this energy must not depend on how we choose our
coordinate system. Hence, like the vectors involved the expression, the shielding
tensor components must obey certain transformation rules under coordinate
transformations.
What are the functions plotted in the nanotube example above?
Clearly, there is a lot of information contained in a rank-2 tensor such as the NMR shielding
tensor .
At some point during our research we felt a need for some kind of graphical
representation of these tensors in relation to the molecular geometry. What we came
up with was the following procedure [94]:
-
Calculate a scalar function
-
Transform
to spherical polar coordinates and divide by
to get
which only depends on the angles
-
Prepare a spherical polar plot of .
A polar plot is done as follows: For a large number of value pairs
within the
allowed ranges of
and ,
in each case a point is created with the coordinate triples
.
The points are then connected to form a surface in three dimensions.
-
In fact, we crate two surfaces, one for all points for which
is positive, and another one for all points for which
is negative. The surfaces are colored differently.
These are the surfaces shown in the nanotube example above, for the NMR shielding
tensors of two different carbon atoms in the nanotube. The blue surfaces are for positive
, the orange ones
for negative .
So what do they represent?
From the NMR nucleus in some direction
,
the distance of the surface from the nucleus (times
depending on the color coding) is equal to the NMR shielding that
would be observed if the external field were in the direction of
relative to a fixed orientation of the molecule. In fact, as my collaborators and I
learned later, such a function had previously been defined as the ‘shielding
response vector’ [1]. The graphics shown above include scaling factors
chosen
to make the polar plots appear in a convenient size relative to the molecule. When the
distance
of the surface from the plot’s origin is multiplied with the scaling factor
one gets the shielding value (i.e. for NMR shielding the scaling factor
is in
ppm per unit distance).
If the shielding is the same no matter what the relative orientation of the field and
the molecule is, then the shielding tensor is said to be isotropic. The corresponding
polar plot is a sphere, the same distance of the surface from the origin in all
directions. Clearly, the shielding tensors of the nanotube are very strongly
anisotropic. The shielding is large & positive for a magnetic field perpendicular to
the sidewall, but very small or even negative for carbons for which the field is
tangential to the sidewall or the caps’ surfaces.
If one performs NMR measurements on molecules that rotate freely, and fast on
the NMR time scale, then the measurement represents an average of all the different
directions of the plot. This isotropic average of the tensor is the isotropic shielding
constant .
This value is a scalar and independent of the chosen coordinate system representing
the shielding tensor.
For a given nucleus in a given molecule, one may ask the question: Is there an
orientation of the molecule such that the Cartesian representation of the shielding tensor
is as
simple as possible?, namely diagonal:
|
|
The answer is ‘yes’, as long as an antisymmetric component, if present, is
subtracted from the shielding tensor. [To my knowledge, an antisymmetric
shielding tensor component is not observed in NMR experiments.] The three
shielding values on the diagonal are the principal components of the shielding
tensor, and the corresponding coordinate system is referred to as the
principal axis system (PAS). What is nice about the polar plot of the tensor
is that the orientation of the PAS is indicated by the extremum values of
,
i.e. by the directions where the surfaces are furthest away or closest to the origin of
the plot. In the nanotube plot above, I have also included the PAS shown as green
and red arrows (green if the principal component is positive, red otherwise),
showing that indeed the polar plot surface indicates the orientation of the
PAS. The principal values are given by the distances of the surface from the
origin in these directions. The nice thing about the polar plots is that if
we rotate the molecule and re-calculate the shielding tensor and prepare
a new polar plot, it will look just the same because the plot depends on
the relative orientation of the PAS and the molecule, and the two rotate
together.
Electric field gradient tensors: We later used such polar plots of tensors extensively
for electric-field gradients (EFGs) [128], which are underlying the quadrupolar
interaction that is so important in solid-state NMR of isotopes with spin
.
For EFG tensors, you would never see a polar plot looking like a sphere
or even a deformed sphere. The reason is that EFG tensors (components
) have
no isotropic part; they are ‘traceless’ or ‘purely anisotropic’ meaning the isotropic
average
is zero. It the PAS, there are then only two unique numbers characterizing the tensor
because the fact that the tensor is traceless fixes the third principal component if two
others are known. Usually, for EFG tensors the principal values are ordered such that
denotes the largest-magnitude diagonal element, and
the
smallest (the signs are not reflected in the ordering). Instead of giving two of the
principal components, the tensor can also be characterized by the values of
and the asymmetry
parameter
(the subscript
indicates this has to do with the nuclear quadrupolar interaction).
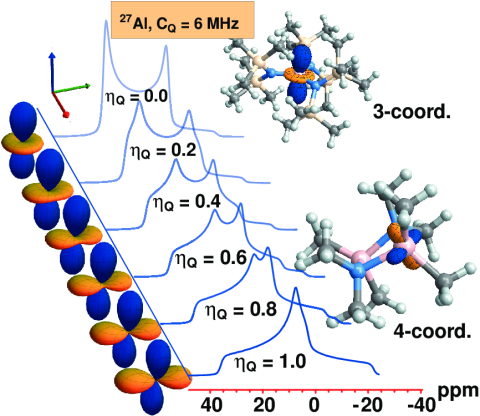
In publication [128] we showed for the quadrupolar
Al
nucleus, how the line shape of a solid-state NMR spectrum changes with the
asymmetry parameter of the EFG tensor. In the figure below, you can see the polar
plots of various EFG tensors and next to them the resulting NMR line shape. This is
from a research project in collaboration with Rob Schurko at Florida State University
(formerly U. of Windsor).
References
[1] Hansen, A. E.; Bouman, T. D. J. Chem. Phys. 1989, 91,
3552-3560.
© 2017 – 2025 J. Autschbach. The material shown on this web page is in parts or
wholly based on the results of research funded by grants from the National Science
Foundation [NSF, grants CHE 0447321, 0952253, 1265833, 1560881, 1855470]
and educational projects supported by these grants. Any opinions, findings,
and conclusions or recommendations expressed in this material are those of
the author and do not necessarily reflect the views of the National Science
Foundation.